
Leber Hereditary Optic Neuropathy (LHON), also known as Leber Optic Atrophy, was named after Doctor Theodore Leber, who described in 1871 a characteristic pattern of sudden vision loss in young men with family history of blindness. It is the most common hereditary optic neuropathy, it is caused by a mitochondrial mutation and has a low prevalence. Although in most geographic areas it is still unknown, it is estimated that it can affect between 1 in every 27,000 to 45,000 people around the world. It affects most usually young men aged between 15 and 25 years old, although it can also affect people from all ages. In women it has a much lower incidence.
It generally causes a sever vision loss in both eyes. In most cases, an eye is first affected, and several weeks or months after, the second eye is affected.
An early diagnosis is key for the evolution of this disease. The probability of a favorable response to a treatment is higher if it is started early. Plus, the diagnosisends the pilgrimage of uncertainty, diagnostic tests and appointments with multiple specialists that these patients usually have to endure and that causes heavy inconvenience for them and their families.
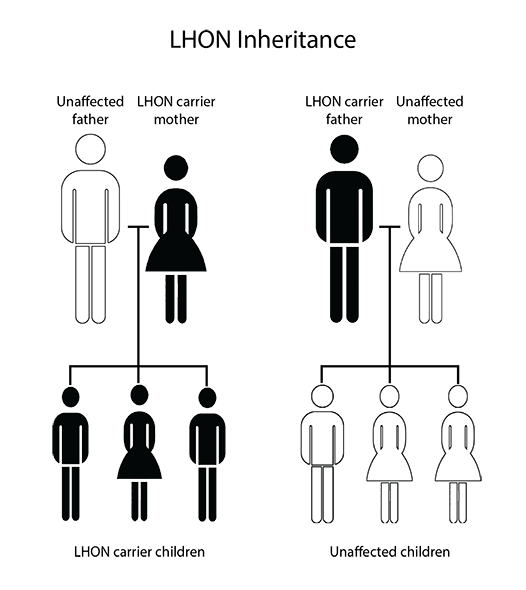
Even though most of our genetic material (DNA) is localized on the cell nucleus, a small part of it is located in mitochondria (mitochondrial DNA).
Genes within the nucleus are inherited from both biological parents. Nevertheless, mitochondrial genes are only inherited from the mother. This means a male with a mitochondrial DNA mutation won’t transmit it genetically to any of his biological sons, while a woman with a mitochondrial DNA mutation will transfer it to all her biological descendants.
More than 95% of LHON cases are caused by one of the three concrete mitochondrial DNA mutations. However, a significant percentage of people who have these mutations never develop the disease symptoms. More specifically, only a 10% of women and 50% of men who are carriers of any of these mutations will develop optic neuropathy. This means other genetic (mitochondrial or nuclear) and/or environmental factors must act in order to trigger the vision loss. For example, exposure to tobacco or high amounts of alcohol in carriers of these mutations increases the risk to develop the disease.
Typically, LHON carriers remain asymptomatic until they experience a loss of vision, more or less sudden, in an eye, and a few weeks or months later, in the other eye.
Vision keeps worsening for some weeks until is deep and, in most cases, central vision (essential for tasks such as reading, driving or recognizing faces) is severely and permanently affected in both eyes, although some people experience some visual recovery after some time. This permanent vision loss is due to the death of optic nerve cells, responsible for sending images from eyes to the brain.
Although vision loss is the only symptom in most LHON patients, arrhythmias, and neurological troubles (postural tremors or other movement disorders) have been described in some of them.
For several reasons, it is a disease with a difficult diagnosis. First of all, it is a hereditary disease, but it does not manifest itself in all people suffering the mutation. For this reason, it may arise with no evidence of a family history of the disease.
In order to diagnose it, it is usually necessary to perform a comprehensive neuro-ophthalmology examination and a blood test that examines the mitochondrial DNA.
Suspect is essential to identify the disease. Here are some of the alarm signs that should make one suspicious of a possible LOHN case:
In an eye fundus examination, there initially might be seen a microangiopathy and a puffiness of the peripapillary nervous fibers layer, which progresses to an optic atrophy.
Currently, the prognosis is usually a permanent and severe vision loss. Several researches are being performed in order to find a treatment for the disease. Dr. Castillo, Head of the Neuro-Ophthalmology Department at ICR, contributed in the LEROS clinical trial as the main researcher.
Lately there has been more awareness about LOHN and the diagnostic time has been reduced. However, in most cases months go by until the diagnostic is suspected.
The low prevalence of this disease and its fast progression make it very necessary that the diagnostic and patient referral criteria is perfectly defined and known by clinicians assisting potential LHON patients.



Contact us or request an appointment with our medical team.


 Request an appointment
Request an appointment